2026-05-06
导读
近日,复旦大学研究团队在国际知名学术期刊《Nature Communications》上发表了题为“Gmppb-mutant mice exhibit dystroglycanopathy symptoms that are rescued with GSK3β inhibition or AAV-mediated GMPPB gene replacement”的最新研究成果。该研究成功构建了高度模拟人类患者症状的GMPPB突变小鼠模型,揭示了GMPPB基因突变通过引发广泛的蛋白低糖基化和抑制Wnt/β-catenin信号通路,进而导致肌肉干细胞分化与再生障碍的病理机制。更为重要的是,研究证实了通过药理学激活Wnt通路或使用AAV介导的基因替代疗法,能够显著改善甚至逆转该疾病的表型,为治疗这类罕见遗传病提供了坚实的临床前证据。
抗肌营养不良型糖基化异常(Dystroglycanopathy, DGP)是类罕见的神经肌肉系统疾病,其共同的病理特征是α-抗肌营养不良聚糖(α-DG)的糖基化异常。其中,GMPPB(GDP-甘露糖焦磷酸化酶B)基因突变是导致该疾病的重要原因之一。GMPPB负责催化合成GDP-甘露糖,这是多种细胞内糖基化途径的核心供体。
然而,由于过去缺乏能够完全重现人类患者临床症状的动物模型,科学界对GMPPB突变如何导致严重的肌肉和神经系统病变知之甚少,临床上也主要依赖糖皮质激素等支持性治疗,缺乏对因的治愈手段。为了打破这一僵局,研究团队构建了多系基因编辑小鼠并开展了深入的机制与治疗探索。
纯合突变导致胚胎致死,P32L杂合突变小鼠再现进行性肌营养不良与神经损伤
研究团队首先基于临床数据找出了最常见且症状严重的GMPPB点突变(如P32L和R287Q),并利用CRISPR/Cas9技术构建了Gmppb敲除(KO)小鼠以及Gmppb P32L和R287Q敲入小鼠模型。
观察发现,Gmppb基因的完全敲除以及P32L纯合突变会导致小鼠在胚胎发育早期(E10.5之前)即发生致死,且这种胚胎致死无法通过孕期补充甘露糖来挽救。然而,携带杂合突变的Gmppb P32L/+小鼠虽然能够存活,却随着年龄增长展现出了与人类患者高度相似的进行性肌营养不良症状。
在32周龄时,Gmppb P32L/+小鼠的肌肉组织中GDP-甘露糖水平显著下降,肌肉纤维出现大小不一、中心核肌纤维比例升高、自噬空泡积累以及线粒体结构扭曲等典型病理变化。除了肌肉病变,这些小鼠还表现出明显的神经系统异常,包括认知功能下降、小脑浦肯野细胞群丢失、坐骨神经外周髓鞘脱失以及神经传导速度显著减慢。
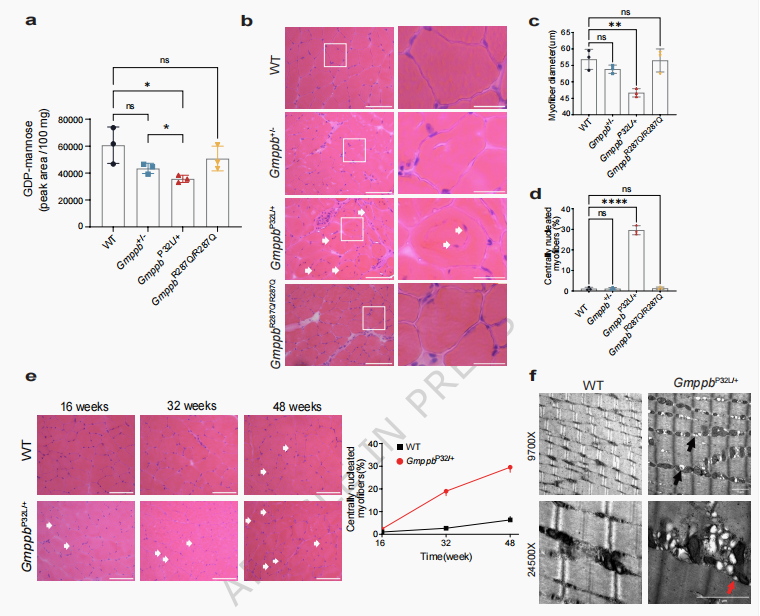
图2 GmppbP32L/+杂合小鼠在骨骼肌中呈现肌营养不良表型
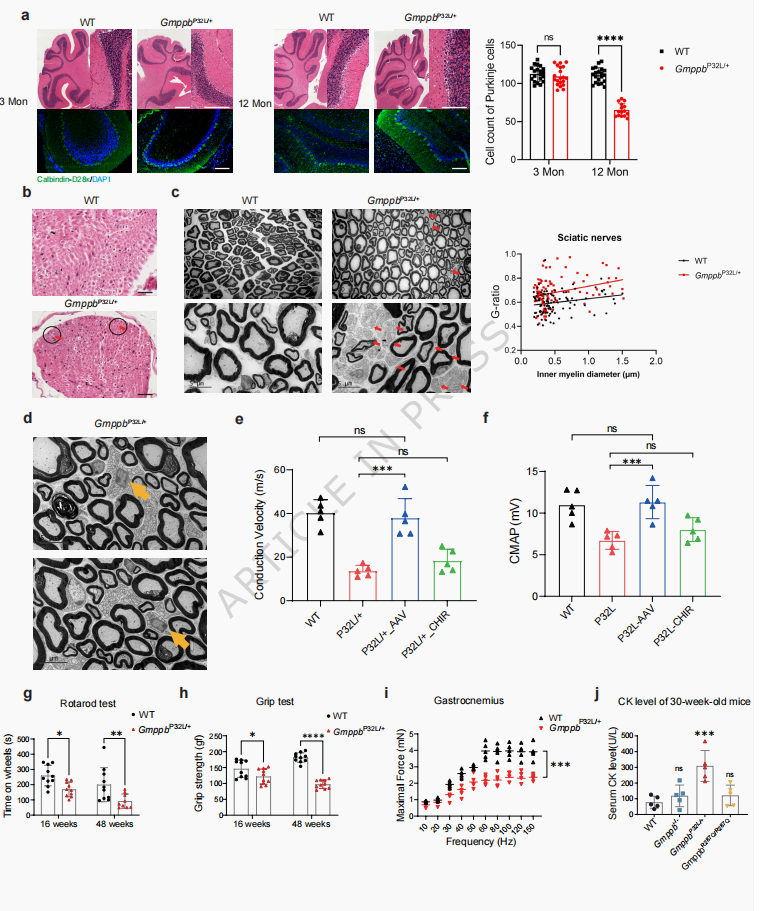
图3 GmppbP32L/+杂合小鼠表现出神经系统受累症状
突变引发广泛的蛋白低糖基化,阻碍肌肉干细胞的分化与再生
由于GMPPB是GDP-甘露糖合成的关键酶,其功能受损最直接的后果就是糖基化异常。研究证实,Gmppb P32L/+小鼠骨骼肌中α-DG的糖基化水平大幅降低,严重削弱了其与细胞外基质(如层粘连蛋白Laminin)的结合能力,直接破坏了肌肉纤维的结构稳定性。
不仅如此,结合转录组学、代谢组学和糖蛋白质组学的多维分析,研究人员发现GMPPB的缺陷不仅局限于α-DG,而是引发了细胞内广泛的蛋白低糖基化事件。这种广泛的糖基化异常引发了连锁反应,导致小鼠体内负责肌肉修复的“后备军”——肌肉干细胞(肌卫星细胞)出现了严重的功能障碍。
在体外分化实验和心脏毒素(CTX)诱导的肌肉损伤模型中,Gmppb P32L/+小鼠的肌肉卫星细胞融合成肌管的能力明显下降,成肌调节因子(如MyoG、MyHC)表达受阻。在损伤后,这些小鼠新生肌肉纤维的横截面积大幅缩小,展现出严重的再生延迟和修复缺陷。
激活Wnt/β-catenin信号通路可有效改善肌肉受损后的再生能力
既然肌肉再生受阻,其背后的分子开关是什么?通过对差异表达基因的深度挖掘,研究团队将目光锁定在了与肌肉分化密切相关的Wnt/β-catenin信号通路上。
在GMPPB缺失的细胞中,经典的Wnt信号通路基因表达显著下调,进入细胞核发挥转录激活作用的活性β-catenin水平也大幅降低。由于多种Wnt受体和配体的稳定性和功能高度依赖于糖基化修饰,GMPPB缺陷引起的全面糖基化异常极有可能“误伤”了Wnt通路。
基于这一机制发现,研究人员引入了GSK3β的小分子抑制剂CHIR-99021。该药物能够绕过上游受体,直接稳定β-catenin蛋白并激活Wnt信号通路。实验结果令人振奋:在给予CHIR-99021治疗后,GMPPB缺失的成肌细胞和突变小鼠的肌卫星细胞的分化和融合能力得到了显著恢复。在活体小鼠肌肉损伤模型中,药物治疗有效增加了再生肌纤维的面积,并在一定程度上恢复了小鼠的运动协调能力和抓力。
AAV介导的GMPPB基因替代疗法全面逆转小鼠运动与神经传导缺陷
虽然药理学激活Wnt通路能够缓解再生障碍,但针对单基因突变引发的罕见病,直接的基因替代疗法(Gene Therapy)往往具有“治本”的潜力。
研究团队采用了嗜肌肉性更强的Myo2A血清型腺相关病毒(AAV),将正常的GMPPB基因通过尾静脉注射导入到12周龄的Gmppb P32L/+小鼠体内。在治疗3个月后,各项生理生化指标均迎来了全面逆转:
组织学与生化逆转:小鼠肌肉中GDP-甘露糖池得到补充,α-DG的糖基化水平恢复正常,肌肉纤维中心核比例显著下降,病理形态明显改善。
运动与肌肉功能:AAV治疗组小鼠在转棒测试中的坚持时间和前肢抓力均大幅提升,甚至达到了与野生型健康小鼠相当的水平;血清肌酸激酶(CK,肌肉损伤标志物)也回落至正常区间。
神经电生理恢复:尤为值得一提的是,直接的基因替换不仅治愈了肌肉,还显著提高了小鼠坐骨神经的神经传导速度和复合肌肉动作电位振幅,有效挽救了神经传导缺陷(这是单独使用CHIR药物无法做到的)。
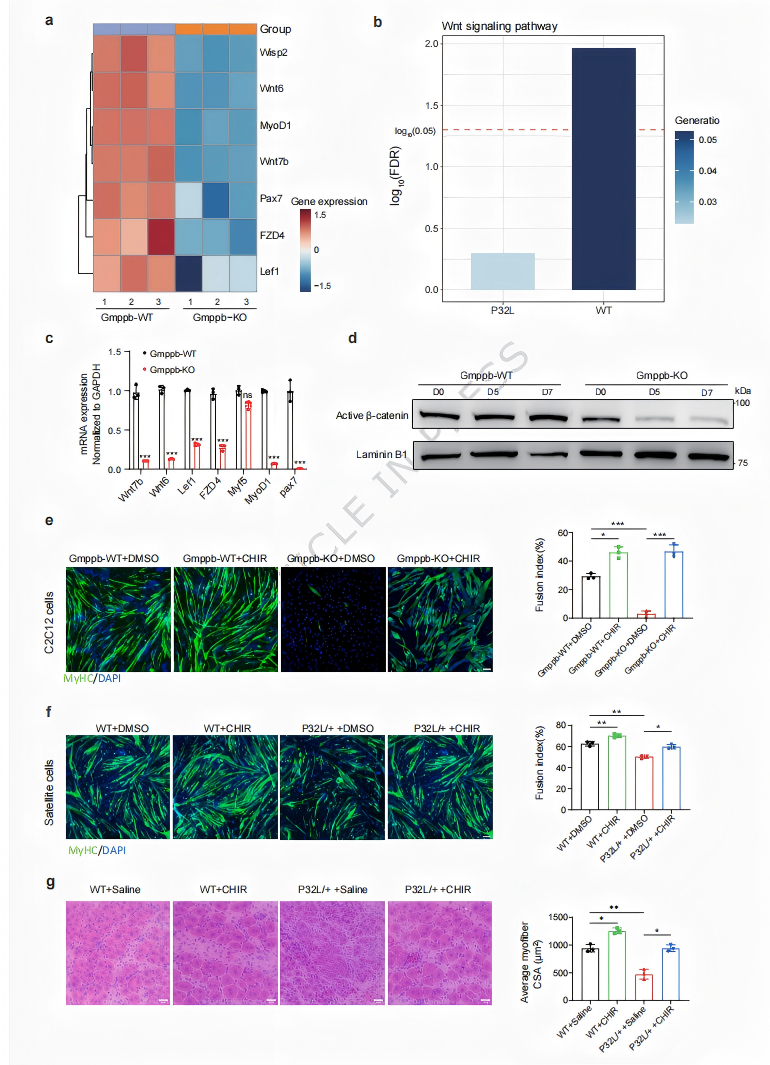
图9 激活Wnt信号通路可挽救肌发生发育障碍
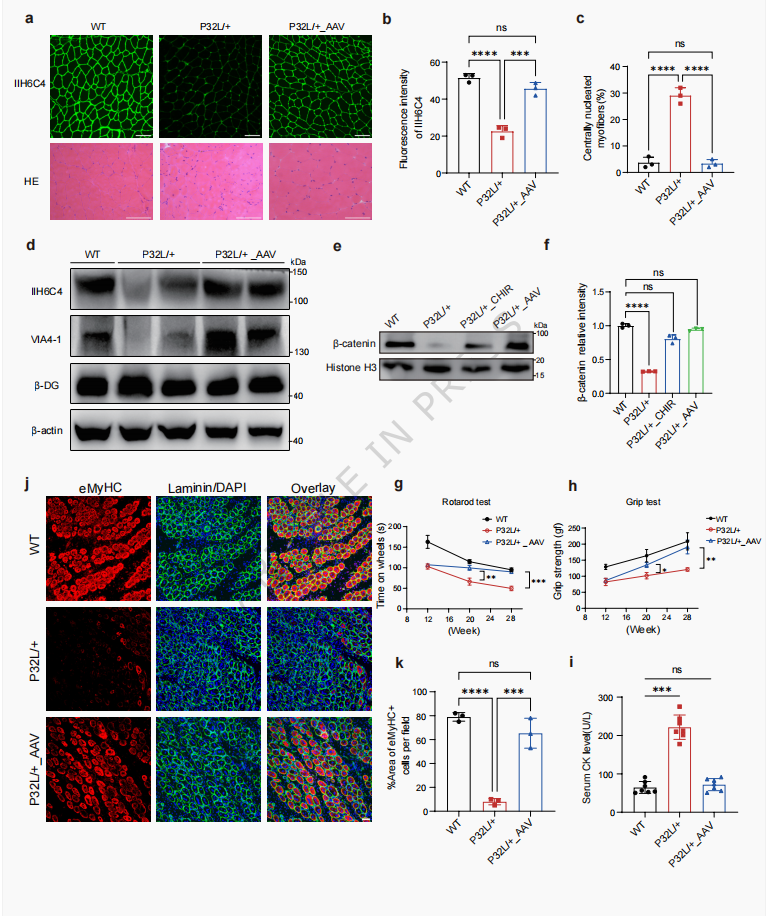
图10 AAV-GMPPB 治疗可改善肌病病理表型
总结
这项历经数年打磨的全面性研究,不仅成功构建了国际上首个忠实还原人类GMPPB相关抗肌营养不良症全貌的杂合突变小鼠模型,更深层次地揭示了“代谢底物缺乏-全面低糖基化-信号通路(Wnt)受阻-干细胞再生失败”这一致病轴心。
更为临床转化指明方向的是,无论是药理学层面的通路干预,还是治本的AAV基因替代疗法,都在活体层面取得了令人瞩目的重塑效果。这不仅为GMPPB突变患者带来了切实的治愈希望,也为其他因代谢酶缺陷导致的先天性糖基化障碍(CDG)疾病提供了通用的研究范式与治疗新思路。

地址:中国武汉东湖高新区光谷七路128号 市场:17720522078 人事行政:027-62439686 邮箱:marketing@genevoyager.com
BD 商务总台:13886000399(BD 经理)
本公司所有产品仅供实验科研使用,不用于人体疾病治疗及临床诊断。
地址:中国武汉东湖高新区光谷七路128号 市场:17720522078 人事行政:027-62439686 邮箱:marketing@genevoyager.com
BD 商务总台:13886000399(BD 经理)
本公司所有产品仅供实验科研使用,不用于人体疾病治疗及临床诊断。
